Chinese Academy of Sciences
Common wheat (Triticum aestivum, AABBDD) is a prime example of an allohexaploid species, having arisen from a series of two sequential hybridization events. This complex genomic structure combines the distinct genetic traits of its three diploid progenitors, endowing the species with considerable genomic plasticity and the capacity for broad environmental adaptation. As a result, Triticum aestivum has been extensively cultivated and is a cornerstone of the global food supply.
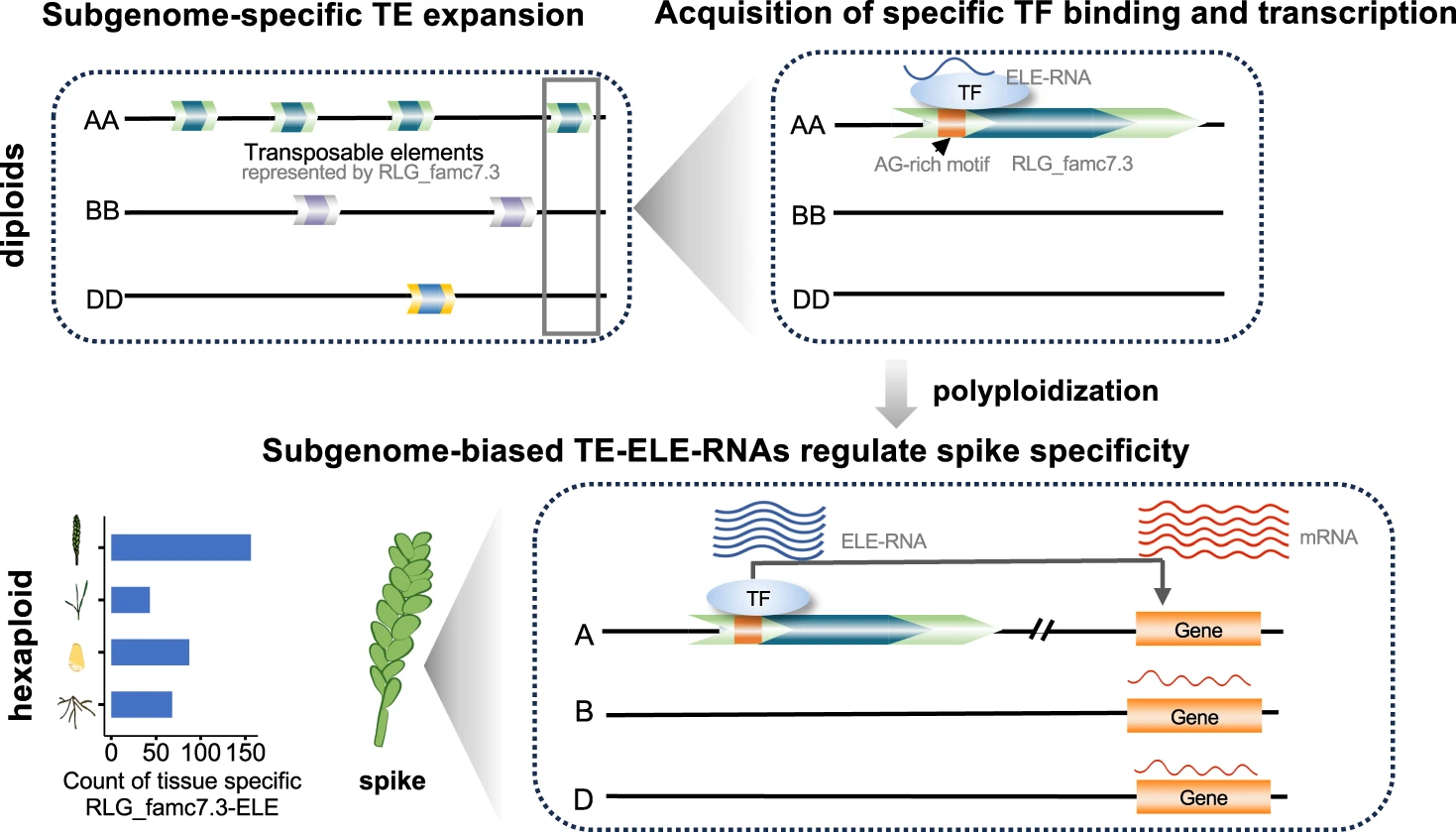
From an evolutionary perspective, the combination of divergent genomes provides a wealth of genetic material that underpins the evolution of polyploids and contributes to phenotypic versatility. However, the intricate molecular mechanisms that drive these processes remain mysterious. Research suggests that the divergence of wheat subgenomes is largely due to species-specific amplification of transposable elements (TEs) in the ancestral diploid species.
Researchers led by Prof. XUE Yongbiao from the Institute of Genetics and Developmental Biology (IGDB) of the Chinese Academy of Sciences, Prof. ZHANG Yijing of Fudan University, Prof. DONG Zhicheng of Guangzhou University, and Prof. Jungnam Cho of Durham University, have developed an innovative computational pipeline using CAGE-seq in conjunction with multidimensional epigenomic datasets to pinpoint the initiation sites of both coding and non-coding transcripts.
Their findings, published in Nature Communications on Nov. 17, reveal a substantial number of nascent strand non-coding RNAs derived from TEs, predominantly situated within the long terminal repeat (LTR) regulatory domains, and exhibiting characteristics of active chromatin states associated with regulatory elements.
Using a combination of high-throughput experimental methods and computational analyses, the researchers uncovered a plethora of distal regulatory elements derived from TEs that exert a significant influence on the transcriptional regulation of subgenomes.
Through a comparative analysis of subgenome expression profiles in different tissues, they identified a TE family with a subgenome-biased amplification pattern that displayed a pronounced tissue-specific expression of nascent strand RNAs. This particular TE family underwent specific expansion in the diploid ancestor of the A subgenome.
Employing RNA interference and overexpression techniques, the researchers showed that aberrant expression of nascent strand RNAs from this TE family induces morphological changes in the wheat spike that may be linked to the domestication of wheat inflorescence architecture.
Although the resulting knockout of TE-derived nascent strand RNAs also induces DNA methylation at the corresponding loci, it remains uncertain whether these phenotypic variations are a direct consequence of TE regulatory element function or are mediated through nascent strand RNAs.
Nevertheless, this investigation posits that the predictive analysis of regulatory element functionality via nascent strand RNAs offers novel methodological paradigms for the functional elucidation of remote regulatory elements within the expansive wheat genome. Specifically, the study provides new insights into the relationship between the abundance of transposable elements and the functional evolutionary dynamics of polyploidy in wheat.
/Public Release. This material from the originating organization/author(s) might be of the point-in-time nature, and edited for clarity, style and length. Mirage.News does not take institutional positions or sides, and all views, positions, and conclusions expressed herein are solely those of the author(s).View in full here.





