Proton hydration, i.e. the involvement of a certain number of water molecules in solvating an excess proton, has been an intensively investigated research topic.
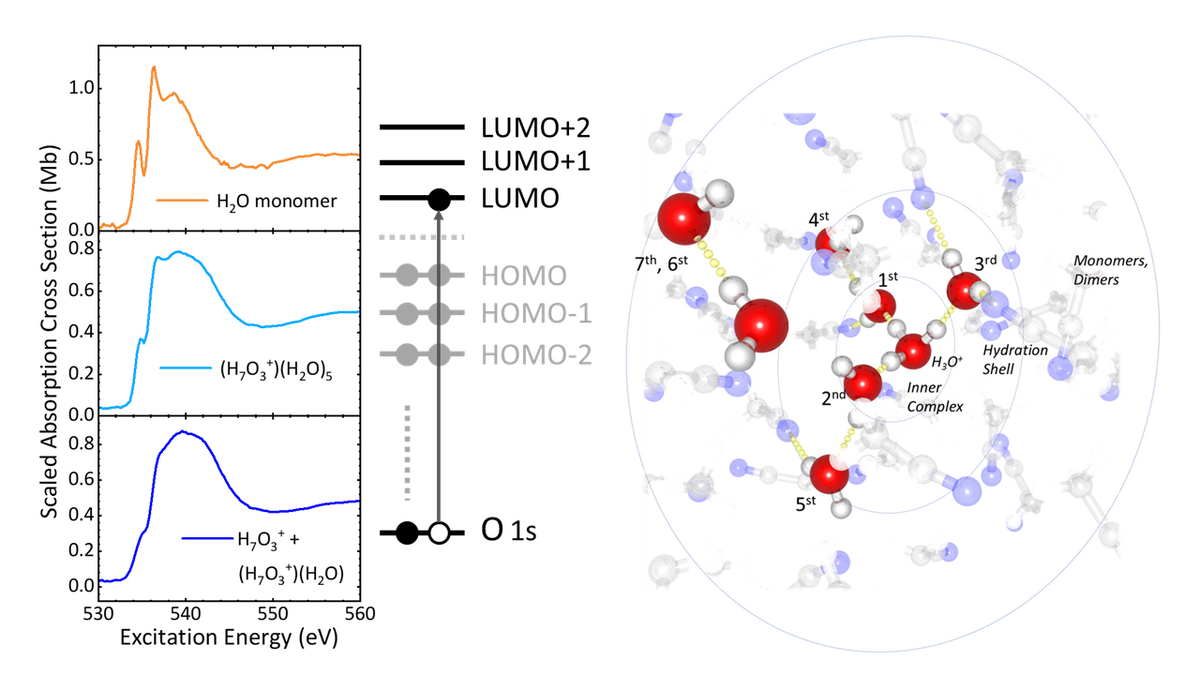
Oxygen K-edge spectra of the water monomer, and of hydrated proton complexes formed by mixing HI and H2O in 1 : 8 and 1 :3.5 mixing ratios respectively, showing the distinct difference in the pre-, main and post-edge band features of monomer water molecules, of hydration shell water molecules and of the inner H7O3+ complex. | MBI
It plays a pivotal role in the mediation (transport) mechanisms of energy conversion and signal transduction, ranging from hydrogen fuel cells to transmembrane proteins. However, aqueous solvation of excess protons has also to date been intensely debated because, being a complex quantum object with strong interactions to its surrounding, it is hard to probe. A collaboration of research teams from the Max Born Institute, the University of Hamburg, Stockholm University, Ben Gurion University of the Negev and Uppsala University have obtained key insight into the electronic structure of hydrated proton complexes in solution, that goes beyond the traditional Eigen or Zundel pictures, where four or two water molecules constitute the hydrated proton complex. Using state-of-the-art liquid flatjet sample delivery technology for measurements of the oxygen K-edge spectra at the BESSYII facility, it has been found that not only the electronic structure of three most inner water molecules in an H7O3+ complex is drastically modified by the proton, but that the first hydration shell around this inner H7O3+ complex, made up by a further 5 water molecules, senses the electric field of the proton by Coulomb interactions.
Textbook chemistry teaches us that bare protons do not exist in aqueous solution, because of the extraordinarily large proton affinity of liquid water. The hydronium ion, H3O+, is what is most familiar: one proton combines with one water molecule into a distinguished own species. In the middle of the 20th century two larger hydrated proton complexes with characteristic well-defined structures have been postulated, one by Manfred Eigen and the other one by Georg Zundel. As an acknowledgement to these pioneering scientists, these have been named after them: the Eigen cation H9O4+ consists of a central H3O+ hydrogen-bonded to three water molecules - H3O+(H2O)3 ; the Zundel cation H5O2+ has a proton shared between two strongly bound water molecules - H2O···H+···OH2. The modern view is that both the Eigen and Zundel ions represent limiting structures of the hydrated proton in liquid water, with the actual structure of protonated water still under debate.
Until now, the electronic structure of hydrated proton complexes under room temperature solution conditions has been elusive, even though the electronic structure would provide vital information on all water molecules that are directly affected by the presence of a proton, providing an electronic structural picture that clearly in its refinement will go beyond the two limiting Eigen and Zundel geometries. Ideally one would need a local probe of electronic charge densities, i.e. a probe such as provided by the electronic orbital structure of the water molecules actively involved in the hydration of the proton. This probe goes clearly beyond a mere determination of local electronic density change from two O-H bonds and two lone pairs, as is the case of the H2O molecule, to three O-H bonds and only one lone pair, as happens for H3O+. In fact, by the formation of hydrogen bonds of different strengths, the electronic orbital structure is expected to change profoundly for all water molecules directly interacting with the proton.
The results obtained on acid/water mixtures dissolved in acetonitrile suggest a general architectural hierarchy for proton hydration, a proton strongly interacting with three water molecules as H7O3+, and one hydration shell affected by the electric field of the positive charge of the proton. This can be concluded from the absence of a distinct pre-edge band and a strong post-edge band in the oxygen K-edge spectrum of H7O3+, whereas water molecules have a clear pre-edge and a much smaller post-edge band. These findings will have direct repercussions in the understanding of proton hydration, ranging from protons in bulk aqueous solution, to proton complexes that are anticipated to occur in hydrogen fuel cell devices, and in hydration pockets in transmembrane protein proton channels. These achievements in soft-X-ray spectroscopy pave the way towards further developments in steady-state and ultrafast soft-X-ray spectroscopy for probing the electronic structure of proton complexes and how elementary steps in proton transport will affect the electronic charge densities accommodating the dislocation of protons.
From Local Covalent Bonding to Extended Electric Field Interactions in Proton Hydration
M. Ekimova, C. Kleine, J. Ludwig, M. Ochmann, T. E. G. Agrenius, E. Kozari, D. Pines, E. Pines, N. Huse, Ph. Wernet, M. Odelius and E. T. J. Nibbering
Angewandte Chemie International Edition 61, e202211066 (2022).





