G protein-coupled receptors (GPCRs) are essential for cell signal transduction and comprise the largest drug target protein family. Upon agonist stimulation, these receptors activate multiple downstream transducers, including G proteins and arrestins, leading to distinct physiological functions. The arrestins play critical roles in modulating GPCR functionalities by terminating G protein signaling and promoting internalization.
Class B GPCRs are involved in many severe diseases such as diabetes, obesity and osteoporosis. These receptors are of great interest as targets for developing biased drugs which selectively trigger the G protein-dependent pathways or arrestin recruitment, aiming for better efficacy and reduced side effects. However, only a few arrestin-bound structures of GPCRs have been determined and all belong to the class A GPCR family. Lack of molecular details of the arrestin engagement at the class B GPCRs limits the understanding of the arrestin-mediated modulation of these receptors and hampers drug discovery.
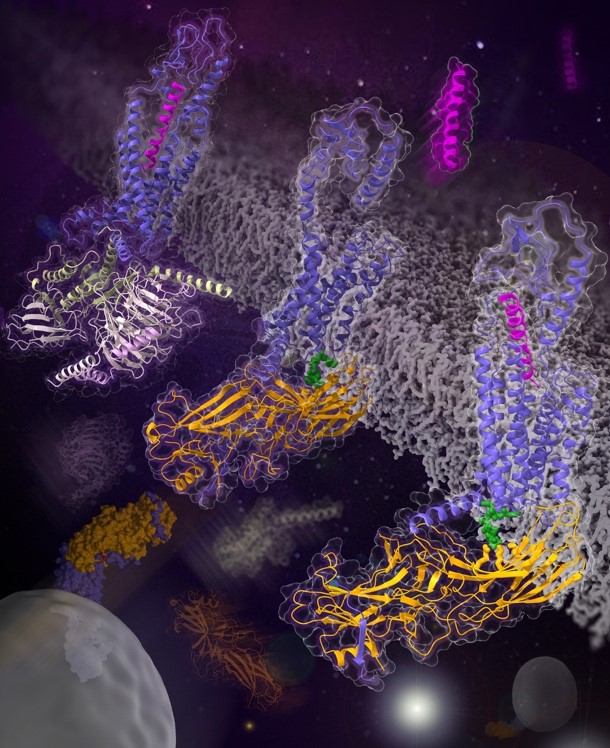
In a study published in Nature, a research group led by WU Beili and ZHAO Qiang at the Shanghai Institute of Materia Medica (SIMM) of the Chinese Academy of Sciences (CAS) determined the cryo-electron microscopy (cryo-EM) structures of the class B glucagon receptor (GCGR) bound to b-arrestin 1 (barr1) in glucagon-bound and ligand-free states. These structures, for the first time, provide a detailed picture of the interaction pattern between a class B GPCR and the arrestin, and unexpectedly reveal many unique features never observed.
The GCGR-barr1 structures display a 'tail' conformation of the complex with the receptor interacting with barr1 mainly through helix VIII in its C-terminal tail. This is in stark contrast to the previously determined GPCR-arrestin structures in which the arrestin adopts a 'core' conformation by binding to both the receptor transmembrane core and the C terminus. The tail-binding pose of barr1 was further defined by a close proximity between the C-edge loops of barr1 and the transmembrane helical bundle in GCGR. In addition, a phosphoinositide derivative bridges barr1 with helix VIII of the receptor to further stabilize the tail engagement.
It was suggested that the tail and core conformations of the arrestin govern distinct processes of receptor signaling and cellular trafficking, and thus result in different cellular responses. The GCGR-barr1 structures, for the first time, offer molecular details of the arrestin coupling to a GPCR in a tail conformation, which greatly extend the knowledge about the mechanism underlying the arrestin-mediated regulation of GPCR signal transduction.
Another striking difference in the GCGR-barr1 structures compared to the previously known arrestin-bound GPCR structures is that the receptor exhibits an inactive conformation even in the presence of the endogenous agonist glucagon. The agonist is either absent or loosely attached to the receptor by binding to a shallower binding site than that in the active GCGR structures.
These structural features are likely attributed to the tail-binding mode of the arrestin, which lacks any contact with the receptor core and does not require the receptor to retain the active conformation. The findings offer a new opportunity for developing novel biased ligands that preferentially recognize different conformations of GCGR with pathway selectivity.
To further investigate the roles of the tail engagement of the arrestin, the scientists performed extensive functional studies using techniques of mutagenesis and bioluminescence resonance energy transfer (BRET). By measuring the arrestin recruitment at the plasma membrane and endocytosis of the wild-type GCGR and its mutants in the binding interface between the receptor and barr1, it was confirmed that the tail conformation of the GCGR-barr complex plays an important role in governing the molecular trafficking of the receptor.