A new computational tool improves the analysis of genetic data, making it easier and faster to study the evolutionary relationships between species.
The shark moth (left) and the figure of eight moth (right), moths belonging to the Family Noctuidae. PsiPartition improved the accuracy of the reconstructed phylogenetic trees for this Family, as well as the bootstrap supports of the branches. (left, Cosmin Manci; right, Ian Peter Morton/Shutterstock)
Researchers at Hokkaido University have developed a new computational tool to help evolutionary biologists analyze complex genetic data to reconstruct the evolutionary history of species and the relationships between them. The tool, called PsiPartition, improves both the computational efficiency of the data analysis and the accuracy of the phylogenetic trees that visualize these relationships. The novel method was published in the Journal of Molecular Evolution.
By analyzing genetic differences between species, evolutionary biologists can infer how closely related they are and map out their evolutionary history. The result of this analysis is a phylogenetic tree, which illustrates the branching patterns of evolution, showing common ancestors and the paths of divergence that led to the diversity of life we see today.
Modern sequencing technologies produce huge amounts of genomic data for phylogenetic analysis, but different regions of the genome evolve at different rates. Some genes evolve more rapidly than others, and specific parts of the genome display unique evolutionary patterns. This phenomenon, known as site heterogeneity, makes it challenging to model evolution accurately using existing approaches, which can be either too slow or imprecise.
"PsiPartition is a useful tool that simplifies the analysis of DNA data by dividing it into groups, or partitions, to account for differences in how fast various parts of the DNA evolve," explains the study's first author, Shijie Xu of the Graduate School of Environmental Science at Hokkaido University. "What makes PsiPartition unique is its ability to quickly and accurately determine evolutionary rates using advanced algorithms. It also automatically identifies the optimal number of partitions to use, saving time and reducing errors common in traditional methods."
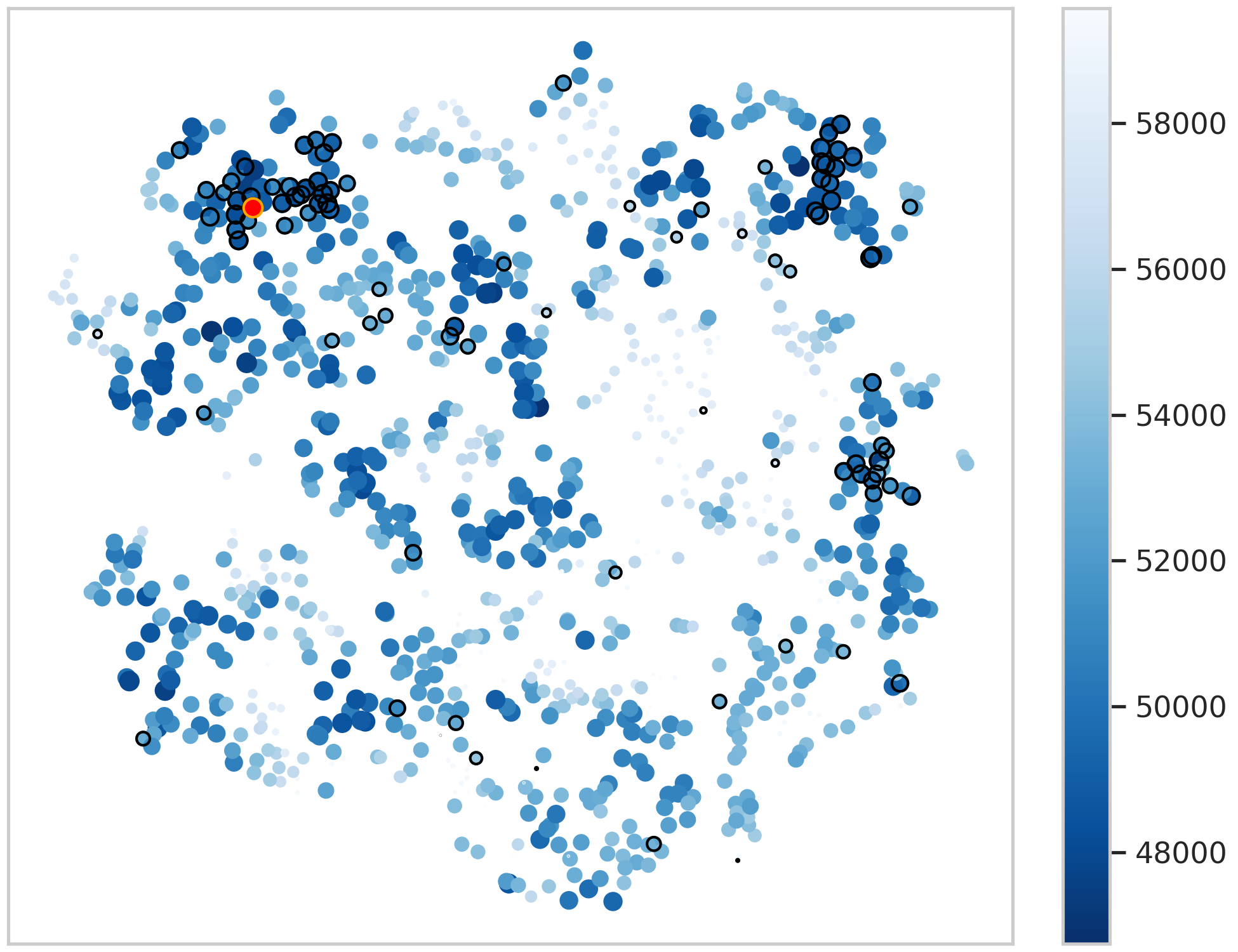
Scatter diagram showing how PsiPartition finds the best model (red spot) for each dataset. (Shijie Xu, Akira Onoda. Journal of Molecular Evolution. December 5, 2024)
PsiPartition delivered impressive results when it was tested with both real and simulated data. It had a significantly improved processing speed, particularly for large datasets, as well as excelling at handling complex, highly variable data. Most notably, for the moth family Noctuidae, it improved the accuracy of the reconstructed phylogenetic trees, as the bootstrap support for the branches was high. Thus, the PsiPartition trees provide a different and possibly more accurate evolutionary reconstruction of these species.

The PsiPartition phylogenetic tree (bottom) for four moth species in Noctuidae, compared to a tree constructed in a different software (top). The PsiPartition tree is different, and possibly more accurate due to the high bootstrap values. Common names of the moths are the shark moth (Cucullia umbricata), the abrupt brother moth (Raphia abrupta or sp), the silver hook moth (Deltote uncula) and the figure of eight moth (Diloba caeruleocephala). (Shijie Xu, Akira Onoda. Journal of Molecular Evolution. December 5, 2024)
"Our tool will allow evolutionary biologists to study species more accurately and efficiently," says lead author Professor Akira Onoda of the Faculty of Environmental Earth Science at Hokkaido University. "By simplifying the analysis of large, complex genomic datasets, PsiPartition provides a powerful new tool for evolutionary research."

Authors Shijie Xu (left) and Akira Onoda (right). (Photo: Shijie Xu)
Original Article:
Shijie Xu, Akira Onoda. PsiPartition: Improved Site Partitioning for Genomic Data by Parameterized Sorting Indices and Bayesian Optimization. Journal of Molecular Evolution. December 5, 2024.
DOI: 10.1007/s00239-024-10215-7
Funding:
This work was supported by Hokkaido University DX Doctoral Fellowship (JPMJSP2119) from the Japan Science and Technology Agency (JST) SPRING; the SATREPS Project "Recovering High-Value Bioproducts for Sustainable Fisheries in Chile (ReBiS)" funded by the JST and the Japan International Cooperation Agency (JICA) (JPMJSA2206); and the Japan Society for the Promotion of Science (JSPS) KAKENHI (JP24H02213) in Transformative Research Areas (A), (JP24A202) Integrated Science of Synthesis by Chemical Structure Reprogramming (SReP), and JP24K01533.





