Chronic inflammation of the pancreas is a debilitating disease with poorly understood causative factors. Now, researchers at Osaka University have identified the disturbed molecular pathways and revealed the underlying mechanisms that may inform an effective and much-needed therapeutic strategy.
The pancreas is an important organ with a dual role in digestion and the production of various hormones including insulin and glucagon that fine-tune blood sugar levels.
Chronic pancreatitis (CP) is characterized by inflammation of the gland causing shrinkage, replacement of glandular elements with fibrous tissue (fibrosis) and loss of function. Patients suffer abdominal symptoms, poor digestion and consequent nutritional disorders. They can also develop diabetes mellitus and, in some cases, even pancreatic cancer. The roles of alcohol abuse and digestive enzyme gene mutations in triggering CP are known, yet the underlying molecular mechanisms remain unclear.
The research team's suspicions fell on two molecular channels of cellular communication, called the PI3K and the Hippo signaling pathways, that have been implicated in the development of pancreatic cancer. Takeshi Tamura, co-first author, explains the research strategy: "In experimental mouse models of CP, we showed that the expression of two molecular components of the PI3K and Hippo signaling pathways, PTEN and SAV1, is decreased in the pancreatic tissues. Additionally, we generated mice that lack pancreas-specific genes for these two molecules and observed that they spontaneously developed CP."
Co-first author Takahiro Kodama elaborates: "In laboratory experiments, we were able to establish the pivotal role of Connective Tissue Growth Factor (CTGF) in the development of CP. Inhibition of PTEN and SAV1 increases CTGF which, in turn, induces a pathological transformation of glandular cell types called acinar-to-ductal metaplasia (ADM). It also activates pancreatic stellate cells and macrophages, both of which promote fibrosis in CP." These are hallmarks of chronic inflammation in the pancreas with a potential to trigger carcinogenesis. Furthermore, the researchers were able to show that CTGF inhibition in animal models alleviated the inflammation, fibrosis and ADM formation of CP, and they could confirm these experimental findings by analysis of human pancreatic tissue.
These findings elaborate the molecular mechanisms that underpin the development and progression of chronic inflammation in the pancreas, suggesting that CTGF may be a fruitful new therapeutic target in the quest for effective therapy against chronic pancreatitis.
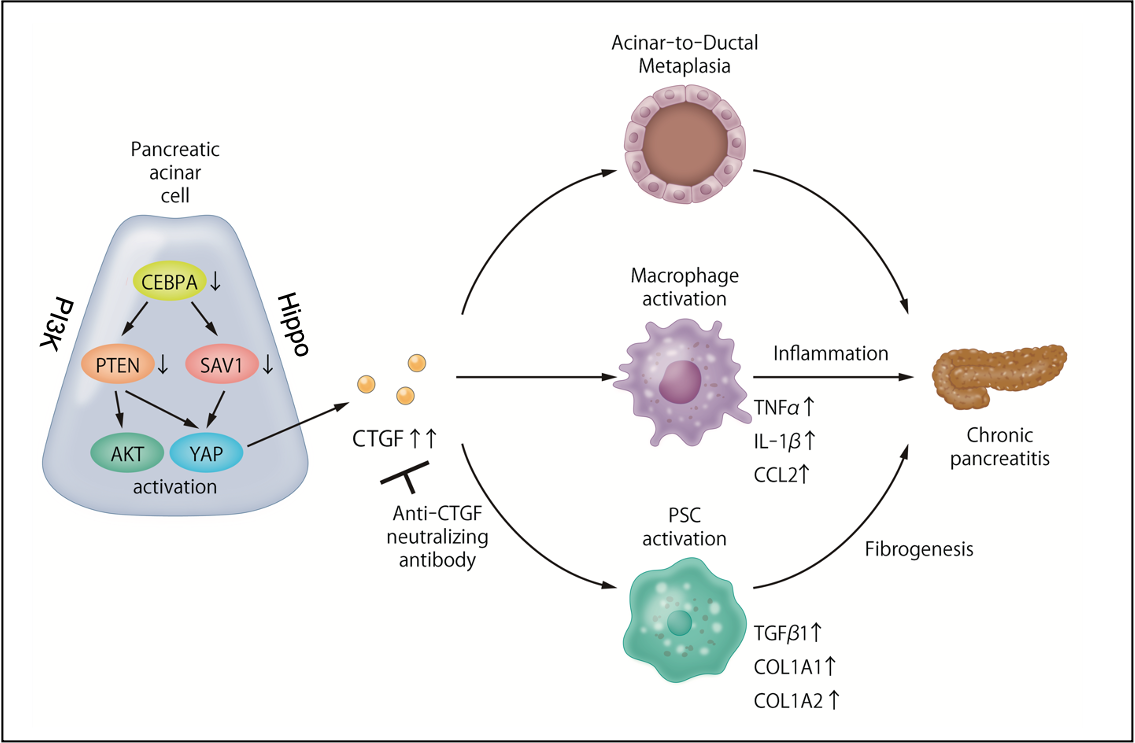
Figure 1. Dysregulation of the PI3K/Hippo signaling pathways is involved in the pathogenesis of chronic pancreatitis, and the resulting overproduction of CTGF is a new therapeutic target for chronic pancreatitis.
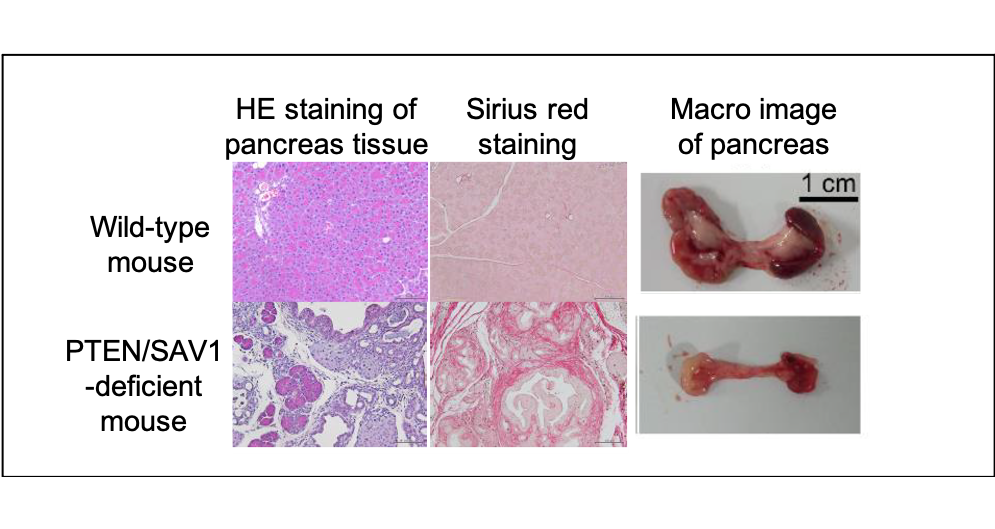
Figure 2. The role of the PI3K/Hippo pathway in chronic pancreatitis.
Mice lacking PTEN/SAV1 spontaneously develop chronic pancreatitis with inflammation, fibrosis and atrophy of the pancreas.
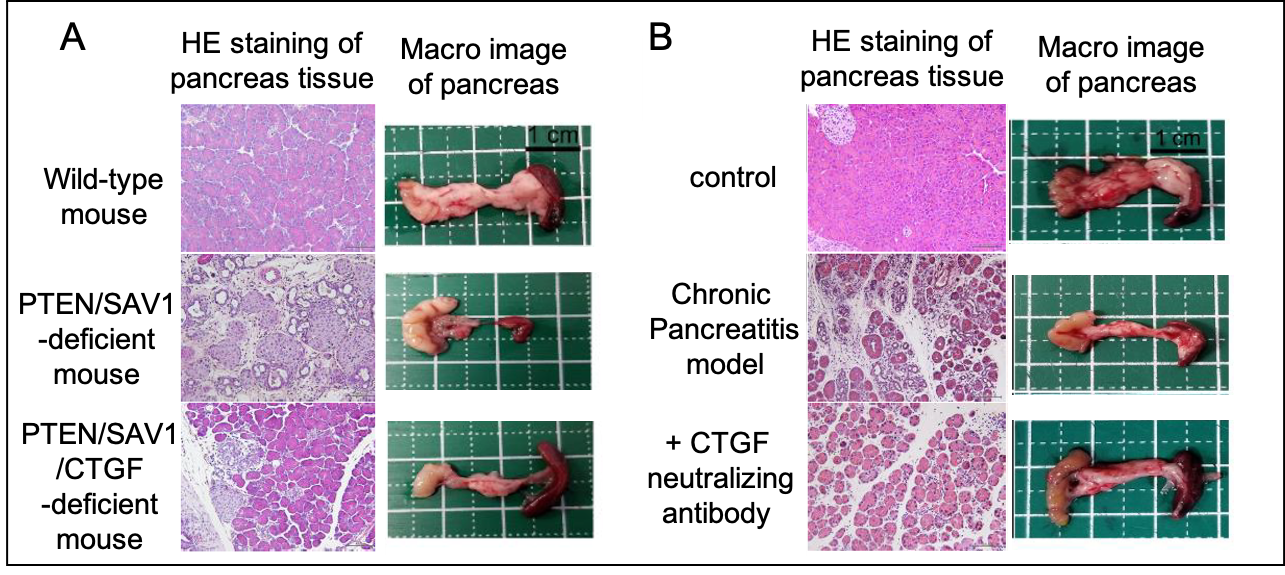
Figure 3. Treatment of chronic pancreatitis by targeting CTGF.
A) Chronic pancreatitis developed in PTEN/SAV1-deficient mice is alleviated by CTGF deficiency.
B) Drug-induced chronic pancreatitis is alleviated by CTGF neutralizing antibodies.
The article, "Dysregulation of PI3K and hippo signaling pathways synergistically induces chronic pancreatitis via Ctgf upregulation" was published in the Journal of Clinical Investigation at DOI: https://doi.org/10.1172/JCI143414.





